Ver traducción automática
Esta es una traducción automática. Para ver el texto original en inglés haga clic aquí
#Novedades de la industria
{{{sourceTextContent.title}}}
Presentación de neoantígenos tumorales
{{{sourceTextContent.subTitle}}}
Presentación de neoantígenos tumorales
{{{sourceTextContent.description}}}
Clasificación de los antígenos tumorales
Los últimos avances en la secuenciación del genoma indican que, durante su inicio y desarrollo, el cáncer adquiere decenas de miles de mutaciones celulares somáticas diferentes. La mayoría de estas mutaciones no confieren ventajas inherentes al crecimiento (mutaciones pasajeras) y suelen ser el resultado de la inestabilidad genómica dentro del tumor.
Un pequeño subgrupo de mutaciones cancerosas altera la regulación celular normal y contribuye a impulsar el crecimiento del cáncer y la resistencia a terapias dirigidas (mutaciones impulsoras). Hasta la fecha, se han identificado aproximadamente 140 genes que pueden impulsar la tumorigénesis. Sin embargo, tanto las mutaciones impulsoras como las pasajeras pueden alterar la secuencia de codificación de aminoácidos, lo que se conoce colectivamente como mutaciones sinónimas, dando lugar a proteínas mutadas que no se expresan en las células normales. Estas secuencias proteicas aberrantes se procesan en péptidos cortos y se unen al complejo mayor de histocompatibilidad (MHC; también conocido como antígeno leucocitario humano HLA en humanos), haciéndolas reconocibles por las células T como antígenos extraños [1].
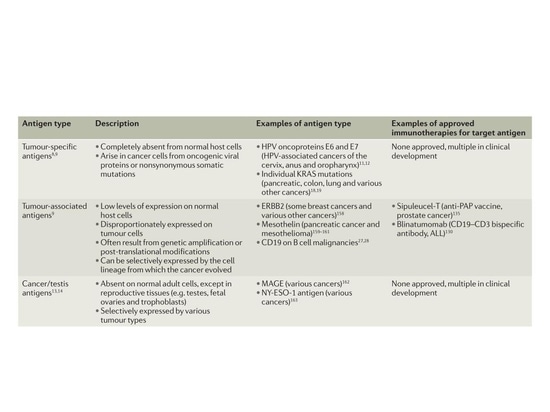
Debido a su expresión selectiva en tumores, los antígenos tumorales específicos (TSA) generados por mutaciones sinónimas y otras alteraciones genéticas se denominan neoantígenos. En los subgrupos de tumores humanos de etiología vírica, como el carcinoma de células de Merkel (CCM) asociado al poliomavirus de células de Merkel (MCPyV) y los cánceres cervicales, orales y de otras localizaciones específicas asociados al virus del papiloma humano (VPH), las proteínas codificadas por el marco de lectura abierto vírico representan otro tipo de neoantígenos. Además de los ATS, existen dos clases principales de antígenos tumorales. Los antígenos asociados a tumores (AAT) se sobreexpresan en las células malignas, pero también se expresan en niveles bajos en las células normales. Los antígenos tumorales/testiculares (ATC) se expresan en varios tipos de tumores y tejidos reproductores (por ejemplo, testículos, ovarios fetales y capas nutricias) pero tienen una expresión limitada en otros tejidos adultos normales y suelen estar ausentes en las células reproductoras normales porque estos tejidos no expresan moléculas de clase CMH. Los neoantígenos pueden producirse a nivel genómico a través de variaciones de un solo nucleótido (SNV), inserciones de bases y fusiones de genes, a nivel transcripcional a través del empalme selectivo, la poliadenilación (pA), la edición del ARN y las denominadas regiones no codificantes, y a nivel proteico a través de la alteración de la traducción y las modificaciones postraduccionales.
▲Clasificación de antígenos tumorales[1]
Clasificación de HLA
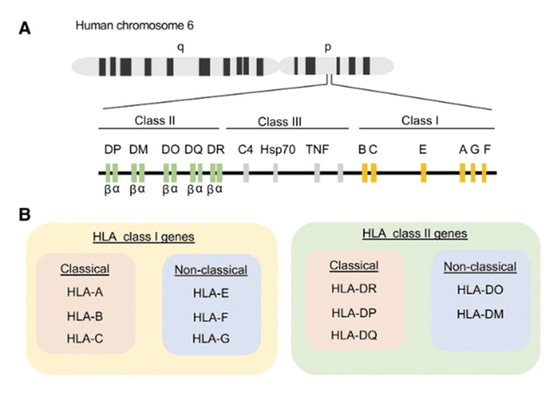
La activación de las células T depende del reconocimiento simultáneo de fragmentos peptídicos extraños y de moléculas del CMH propias, un fenómeno conocido como restricción del CMH. Las células T CD8+ están restringidas por el CMH-I, mientras que las células T CD4+ lo están por el CMH-II. El complejo mayor de histocompatibilidad (CMH), denominado antígeno leucocitario humano (HLA) en humanos, está codificado en el cromosoma 6 del genoma humano. Se trata de un complejo génico altamente polimórfico que codifica moléculas de la superficie celular especializadas en la presentación y el reconocimiento de péptidos propios y ajenos. El HLA se divide en tres clases basadas en su función y estructura: HLA-I, HLA-II y HLA-III. Las moléculas de clase HLA se expresan en la superficie de las células nucleadas, excluyendo las células germinales y algunas células neuronales. A diferencia de las moléculas de clase HLA-I, las moléculas de clase HLA-II suelen encontrarse en células presentadoras de antígenos (APC) profesionales, como células B, macrófagos, células dendríticas, células de Langerhans, epitelio tímico y células T activadas (en lugar de en reposo). La estructura y función de las moléculas de clase HLA-III no se conocen bien, pero se sabe que participan en el proceso inflamatorio sin unirse directamente a antígenos.
El HLA se divide a su vez en genes clásicos y no clásicos. Los genes HLA-I clásicos incluyen HLA-A, HLA-B y HLA-C, mientras que los alelos no clásicos incluyen HLA-E, HLA-F y HLA-G. Los genes HLA-II clásicos incluyen HLA-DR, HLA-DP y HLA-DQ, mientras que los alelos no clásicos incluyen HLA-DO y HLA-DM. Los linfocitos T CD8+ humanos reconocen los péptidos presentados por HLA-A y HLA-B clásicos y, en menor medida, por HLA-C. Las células T CD4+ humanas reconocen los péptidos presentados por HLA-DR, HLA-DQ y HLA-DP. El HLA-I consta de tres dominios extracelulares (α1, α2 y α3) unidos de forma no covalente a una molécula de β2-microglobulina. El HLA-II es un heterodímero compuesto por una cadena α y otra β. Los dominios extracelulares del HLA forman una hendidura de unión al antígeno, compuesta por dos hélices α que rodean láminas plegadas β antiparalelas. Esto crea una plataforma capaz de alojar un segmento corto de aminoácidos (aa) denominado péptido. Estos péptidos se unen al fondo del surco de unión mediante interacciones con aminoácidos específicos (denominados residuos de anclaje). Debido a la estructura cerrada de la hendidura de unión del HLA-I, éste suele unir péptidos pequeños de 8-10 aminoácidos, mientras que el HLA-II puede unir péptidos más largos que superan los 11 aminoácidos.
▲Clasificación de los gliomas difusos en adultos[2]
Presentación de los neoantígenos
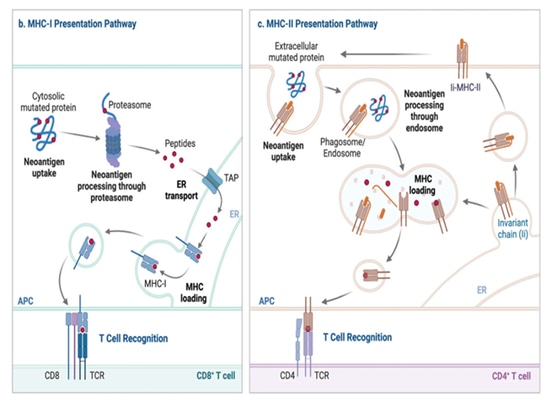
La presentación de proteínas MHC-I se origina principalmente a partir de péptidos del interior de la célula. Durante la homeostasis, las proteínas celulares son degradadas por los proteasomas, generando pequeños péptidos. Los proteasomas son complejos multiproteicos que degradan las proteínas en pequeños fragmentos peptídicos. Durante la infección vírica, la acción de los interferones induce la formación de un complejo proteasoma alternativo denominado inmunoproteasoma, que potencia la generación de péptidos representados por el CMH-I. En consecuencia, al sintetizar proteínas víricas, éstas son el objetivo de la degradación por el inmunoproteasoma, ya sea como proteínas totalmente plegadas o como productos ribosómicos defectuosos. El resultado es la producción de pequeños fragmentos peptídicos derivados del virus infectante, que pueden ser modificados por aminopeptidasas citoplasmáticas.
A continuación, estos fragmentos peptídicos son transportados al retículo endoplásmico (RE) por un mecanismo de transporte de proteínas conocido como proteína de transporte relacionada con el procesamiento de antígenos (TAP). En el RE, los péptidos sufren una modificación adicional por parte de la aminopeptidasa 1 del retículo endoplásmico (ERAP1) si son demasiado largos para la unión al CMH-I tras la translocación a través de la TAP. En el RE, las moléculas MHC-I vacías se asocian con complejos de carga de péptidos (PLC), que incluyen proteínas chaperonas como Tapasin y Calnexin. El PLC mantiene el MHC-I vacío en una conformación receptiva a los péptidos, y cuando éstos son translocados por el TAP, esta conformación facilita la unión al péptido. TAP también forma parte de PLC. La unión del péptido estabiliza la proteína MHC-I, liberándola de los socios de control de calidad del retículo endoplásmico, y es transportada a la superficie celular a través del aparato de Golgi.
Este proceso permite a las células T CD8+ buscar signos de infección o malignidad en el repertorio proteico celular. Sin embargo, en subconjuntos específicos de células dendríticas existe una vía alternativa, conocida como presentación cruzada, que implica la captación de antígenos extracelulares, su transferencia retrógrada desde los fagosomas al citoplasma y su posterior degradación para la carga del CMH-I en los proteasomas y el RE.
▲Vías de producción y presentación de neoantígenos[3]
Los seres humanos tienen más de 24.000 alelos diferentes del antígeno leucocitario humano de clase I (HLA-I, que incluye HLA-A, -B y -C) y de clase II (HLA-DR, HLA-DQ y HLA-DP), lo que da lugar a un polimorfismo diverso. La combinación de estos alelos contribuye a la diversidad del polimorfismo. Los alelos HLA del paciente determinan su repertorio de neoantígenos específicos del tumor, que se presentan para su reconocimiento por las células T. Además, la pérdida de heterocigosidad HLA (HLA-LOH), que se produce en el 40% de los cánceres de pulmón de células no pequeñas, deteriora la presentación del neoantígeno, promoviendo la evasión inmunológica. Por lo tanto, uno de los pasos iniciales cruciales en la predicción de neoantígenos es identificar el genotipo HLA del paciente. Actualmente pueden aplicarse varios métodos computacionales a los datos de secuenciación de próxima generación (NGS) para lograr este objetivo.
Estrategias terapéuticas basadas en neoantígenos
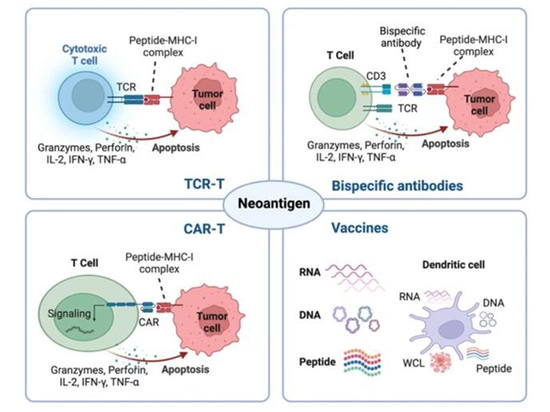
Debido a la ausencia de selección tímica y tolerancia central, los neoantígenos tumorales específicos generados por alteraciones genéticas provocan células T de alta afinidad. Aprovechando su especificidad tumoral e inmunogenicidad, los neoantígenos sirven como dianas emergentes para la inmunoterapia del cáncer, incluyendo vacunas tumorales, terapias celulares adoptivas (ACT), tratamientos basados en anticuerpos y potenciales predictores de inhibidores de puntos de control inmunitario (ICB) [3].
características moleculares del glioma difuso de alto grado en niños[3]
El nuevo antígeno se compone de nuevos antígenos personalizados dirigidos específicamente a cada paciente o de nuevos antígenos comunes expresados en muchos pacientes con cáncer. Las terapias basadas en antígenos nuevos comunes consumen más recursos y tiempo que las terapias con antígenos nuevos personalizados. Dado que los nuevos antígenos personalizados son específicos para cada paciente, no pueden utilizarse para tratar a un gran número de pacientes. Con los últimos avances en secuenciación de alto rendimiento, los nuevos antígenos personalizados permiten al sistema inmunitario dirigirse a epítopos inmunogénicos de tumores malignos sin antígenos comunes predefinidos.
Relación entre TMB y TNB
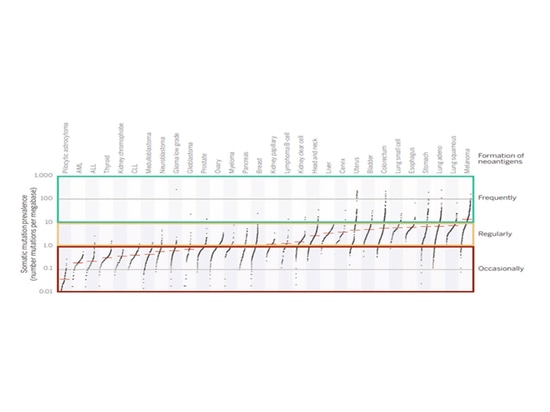
En la mayoría de los pacientes con melanoma, la TMB es superior a 10, lo que conduce a la generación de neoantígenos más eficaces. Basándose en estos datos, se puede predecir que si la TMB es superior a 10 en el tumor, se producirá un número considerable de neoantígenos. Si la TMB está entre 1 y 10, aún existe la posibilidad de portar neoantígenos. En general, si la TMB es inferior a 1 en el tumor, suele ser difícil generar neoantígenos reconocibles por las células T. Los tumores con un TMB elevado (>10) indican que hay más neoantígenos tumorales en la superficie de las células tumorales, lo que provoca una destrucción más eficaz por parte de las células inmunitarias. Además, los pacientes con un TMB elevado pueden presentar mejores respuestas al tratamiento con inhibidores de puntos de control inmunitarios.
▲La TMB tumoral y el potencial para producir neoantígenos[4]
Los retos de la aplicación clínica
1. La inmunoterapia basada en nuevos antígenos sólo muestra una eficacia objetiva en un pequeño subconjunto de respuestas de pacientes bien documentadas. Por lo tanto, se necesitan mejoras significativas para mejorar los resultados clínicos, incluyendo el aumento de la precisión de la predicción de nuevos antígenos, la superación de la evasión inmune y la optimización de la tubería para el proceso de producción.
2. Precisión limitada de la predicción de nuevos antígenos: La aplicación generalizada de la inmunoterapia personalizada se ve restringida por el descubrimiento limitado de nuevos antígenos específicos del cáncer, debido a la heterogeneidad de las cargas de mutación y a las diferencias significativas en la presentación de nuevos antígenos entre los distintos tipos de tumores. Sólo el 10% de las mutaciones no sinónimas de las células tumorales pueden generar péptidos mutados con alta afinidad por el CMH, y sólo el 1% de los péptidos unidos al CMH son reconocidos por las células T de los pacientes.
3. Pérdida de nuevos antígenos: La ausencia de nuevos antígenos específicos del tumor puede ser una estrategia de evasión inmunológica crucial para los tumores. La pérdida de nuevos antígenos puede inducirse a través de varias vías, como la pérdida del número de copias, la supresión transcripcional, el silenciamiento epigenético y los mecanismos postraduccionales.
4. Producción insuficiente de nuevas células T antígeno-específicas: El producto de detección de genes Panorama 602 de Foshu Bioscience incluye detección de pérdida de heterocigosidad HLA-I para evaluar los beneficios de la inmunoterapia adyuvante. También incluye información sobre terapias dirigidas, quimioterapia, carga mutacional tumoral (TMB), carga de neoantígenos tumorales (TNB), factores inmunológicos positivos y negativos, y otra información relevante sobre el uso de fármacos, subtipificación, pronóstico, genética, etc., para proporcionar a los pacientes el máximo beneficio posible.
Referencias
1. Nat Rev Cancer. 2017 Abr;17(4):209-222.
2.Viral Immunol. 2020 Abr;33(3):160-178.
3.Signal Transduct Target Ther. 2023 Jan 6;8(1):9.
4.Science. 2015 Abr 3;348(6230):69-74.
Declaración: Este artículo es solo para compartir. Si hay algún problema de derechos de autor, póngase en contacto con nosotros lo antes posible y lo corregiremos lo antes posible. Gracias